PFIC results in disrupted hepatocyte and/or intrahepatic bile duct function1
Commonly presents in early infancy but can manifest later in life3
Spectrum of rare autosomal recessive genetic disorders involving the liver1
PFIC occurs in an estimated 1 out of every 50,000 to 100,000 births worldwide2
PFIC is progressive in nature and there are several types
Types 1-3 are the most common4
Other types of PFIC exist, with varying phenotypes5
All types of PFIC can be characterized by abnormal bile flow (cholestasis)5
All are progressive in nature6
At the root of PFIC is cholestasis, a dysfunction of enterohepatic circulation
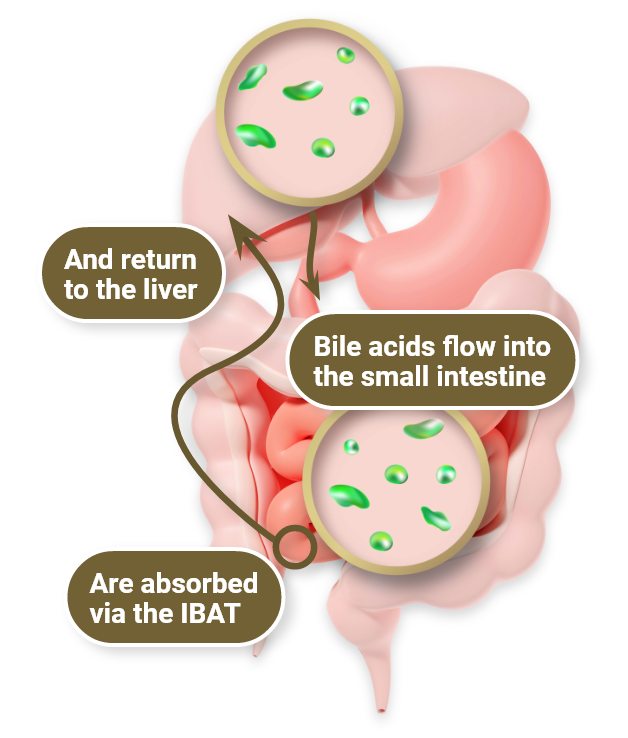
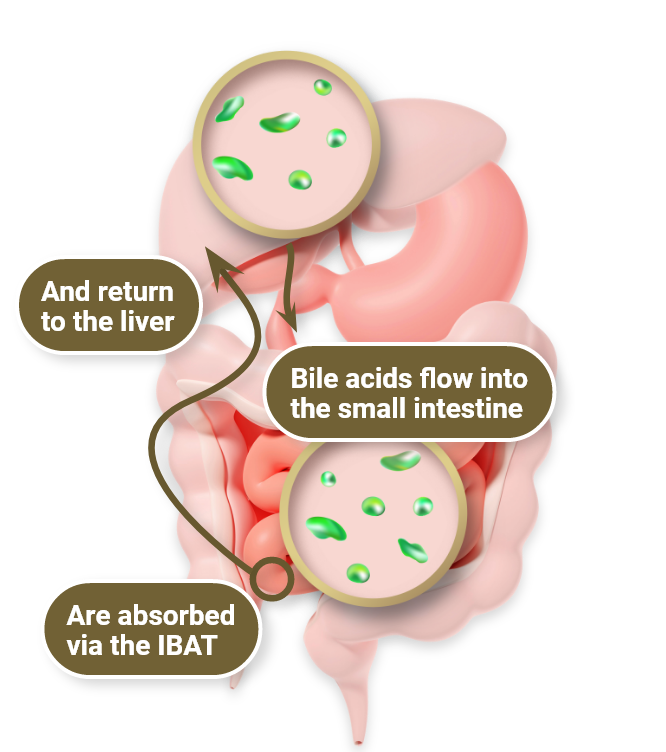
In physiologic enterohepatic circulation1
- Bile acids are synthesized in liver cells and secreted into bile, primarily via the BSEP
- Most bile acids are reabsorbed in the terminal ileum via the IBAT and return to the liver via portal circulation
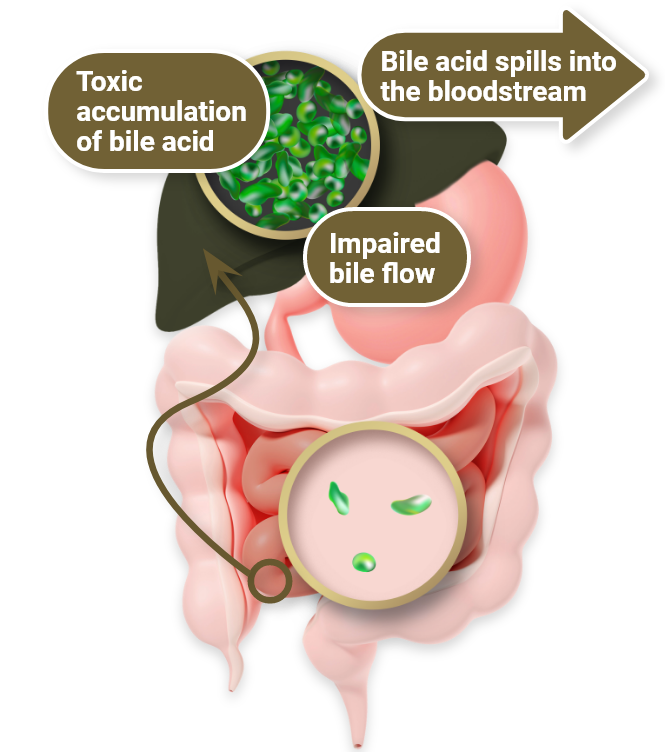
In cholestasis1,7
- Healthy enterohepatic circulation is disrupted by an impairment in bile flow from the liver to the intestines
- This leads to a toxic accumulation of bile acids as they build up in the liver, damaging tissues
- Excess bile acids also “spill over” into the bloodstream, elevating sBA
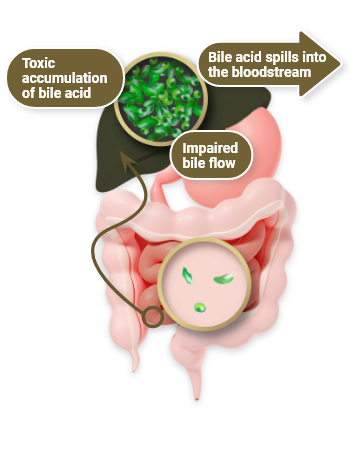
PFIC is generally caused by a genetic defect linked to bile secretion. Though the exact defect varies slightly depending on PFIC type, all types cause cholestasis1,5
Pruritus is a debilitating and challenging symptom of PFIC that can lead to many consequences, including3,8

- Disrupted school activity
- Cutaneous mutilation, scarring
- Emotional distress
- Sleep deprivation
- Impact on caregiver’s personal and family activities
- Impact on caregiver’s employment and finances
The debilitating nature of cholestatic pruritus may necessitate transplantation9
Risks of surgical intervention1,9
While patients with cholestatic liver disease like PFIC can progress to needing lifesaving surgical intervention like SBD or liver transplant, these options carry significant morbidity and mortality
- Invasive approach
- Potential risk for graft rejection
- Need for lifelong immunosuppression

Explore a non-surgical treatment for cholestatic pruritus in patients with PFIC
BSEP=bile salt export pump; ChLD=cholestatic liver diseases; IBAT=ileal bile acid transporter; PFIC=progressive familial intrahepatic cholestasis; sBA=serum bile acid; SBD=surgical biliary diversion.
References:
- Kamath BM, Stein P, Houwen RHJ, Verkade HJ. Potential of ileal bile acid transporter inhibition as a therapeutic target in Alagille syndrome and progressive familial intrahepatic cholestasis. Liver Int. 2020;40(8):1812-1822.
- Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009;4:1.
- Srivastava A. Progressive familial intrahepatic cholestasis. J Clin Exp Hepatol. 2014;4(1):25-36.
- Baker A, Kerkar N, Todorova L, Kamath BM, Houwen RHJ. Systematic review of progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2019;43(1):20-36.
- Goldberg A, Mack CL. Inherited cholestatic diseases in the era of personalized medicine. Clin Liver Dis (Hoboken). 2020;15(3):105-109.
- Amirneni S, Haep N, Gad MA, Soto-Gutierrez A, Squires JE, Florentino RM. Molecular overview of progressive familial intrahepatic cholestasis. World J Gastroenterol. 2020;26(47):7470-7484.
- Pollock G, Minuk GY. Diagnostic considerations for cholestatic liver disease. J Gastroenterol Hepatol. 2017;32(7):1303-1309.
- Mighiu C, O’Hara S, Ferri Grazzi E, et al. Impact of progressive familial intrahepatic cholestasis on caregivers: caregiver-reported outcomes from the multinational PICTURE study. Orphanet J Rare Dis. 2022;17(1):1-9.
- Kamath BM, Baker A, Houwen R, Todorova L, Kerkar N. Systematic review: the epidemiology, natural history, and burden of Alagille syndrome. J Pediatr Gastroenterol Nutr. 2018;67(2):148-156.